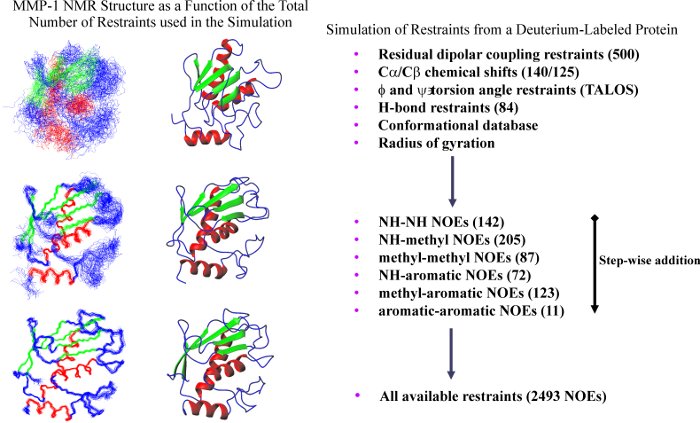
The extensive time-commitment required to solve a protein structures by traditional NMR techniques necessitates alternative approaches to expedite the process, especially in structure-based drug design projects. The utilization of a minimal set of diverse structural information to determine an initial protein fold may prove to be a viable approach and as a starting point for the full refinement of the proteins structure.
The application of deuterium labeling and residual dipolar coupling constants in combination with other structural information has demonstrated the potential for significantly expanding the range of viable protein targets for structural analysis by NMR. A previous study by Clore et. al (1999) demonstrated that a significant improvement in the overall protein structure occurs with the combination of residual dipolar coupling constants and minimal tertiary long-range distance restraints. The analysis of NMR protein structures determined with minimal structural information is extended with a particular interest in the utility of these structures for a structure-based drug design program. As an example, the catalytic fragment of human fibroblast collagenase (MMP-1) was used to follow the effect of minimal restraint sets on the protein structure and its utility in drug design with a particular interest in the effect on the active site conformation.
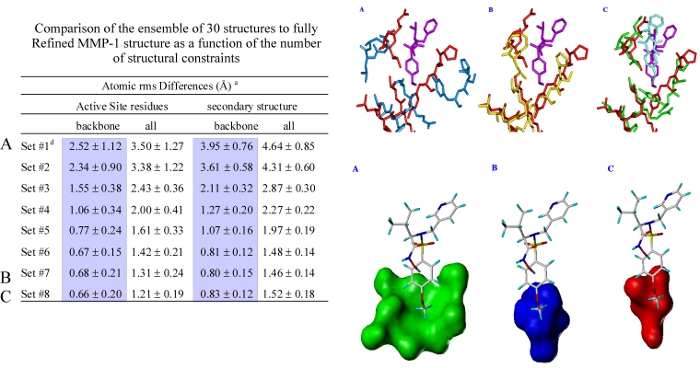
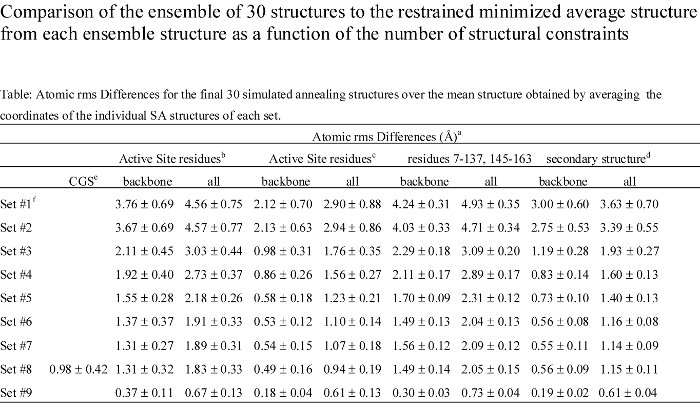
An MMP-1 structure that was calculated with the maximal number of restraints attainable with the constraint of a deuterated protein was shown to be very similar to a high-quality MMP-1 structure that was calculated from a complete set of restraints. The superposition of the active sites backbone atoms for the high-quality and minimal restraint MMP-1 structures yielded an rms of 0.68 Angstroms where the size and shape of the S1' pocket is nearly identical. Additionally, an MMP-1:CGS-27023A complex based on a minimal set of NOE based restraints reliably reproduced the structure of the complex establishing the usefulness of the structures for drug design.
To no surprise, as more structural constraints are added to the calculations, the quality of the structures improves. But, the structural improvement is asymptotic, where the improvement quickly levels off after a few hundred NOE distance constraints.

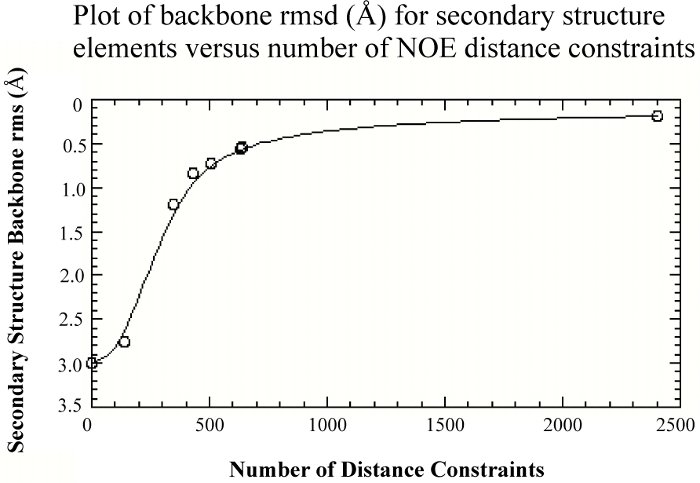
Thus, it is clearly beneficial for the initial determination of a protein fold to maximize both the number and nature of constraints used to calculate the structure. Towards this end, we looked at the validity of using radius of gyration constraints in structure calculations. A recent study has shown that incorporating the radius of gyration (Rg) as a structure restraint target function can improve the packing and accuracy of a structure calculated by NMR. It has also been proposed that NMR structures tend to be poorly packed relative to X-ray structures. Since the overall fold of a protein determined by NMR is primarily a result of the experimental NOE distance restraints, the resulting protein packing is presumably determined from the balance of the NOE distance restraints and the repulsive forces from the dynamics force field function. Thus, if the number of repulsive interactions from the dynamics force field overwhelms the number of experimental restraints the resulting structure may be biased towards an expanded structure.
Based on our analysis, the utilization of the radius of gyration target function as a routine component of an NMR structure determination protocol appears to be a valid approach and does not appear to bias the resulting structure. This is supported by the observation that a number of NMR protein structures exhibit very similar Rg values compared to the corresponding X-ray structure and Rg(pred). Also, this result provides support for the validity of using Rg(pred) in a structure calculation, which is critical given the observation that experimental Rg values over-estimate the structural radius of gyration. The comparison of the Rg values calculated from X-ray and NMR protein structures suggest that NMR structures, in general, are not expanded relative to X-ray structures. The use of the radius of gyration could prove to be a valuable addition to the structure determination of large molecular weight proteins where minimal restraint information may be available.
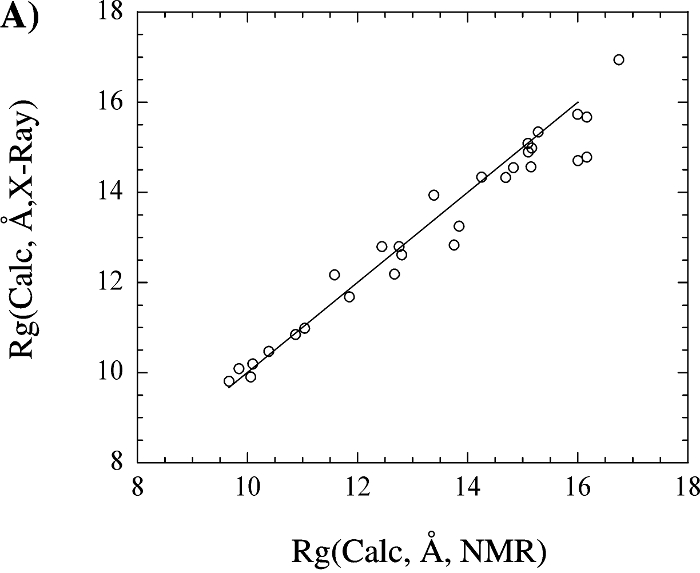
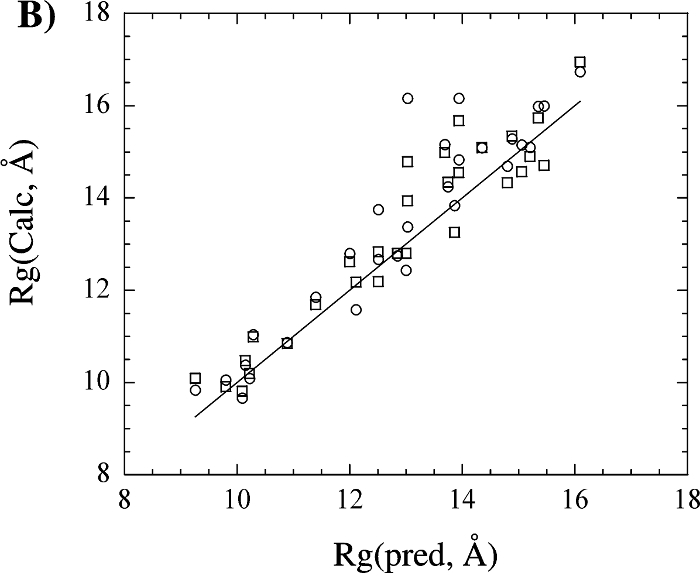
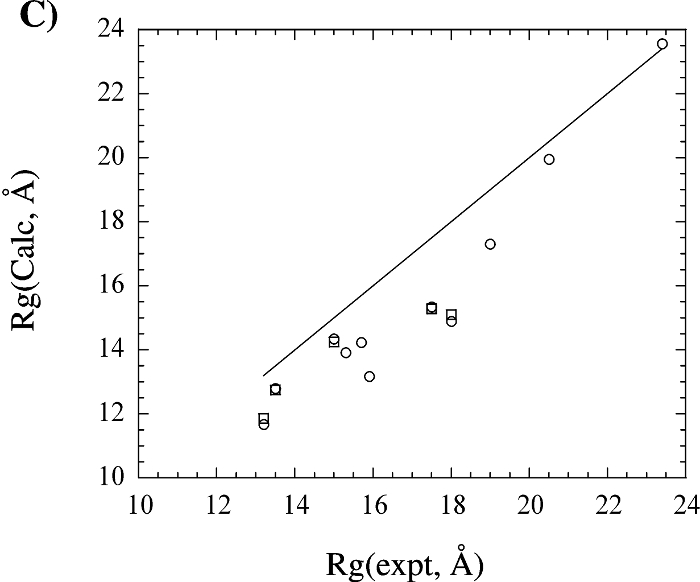
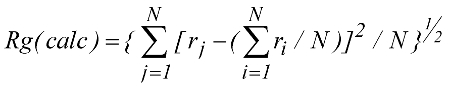
The above figure shows (A) a comparison of the radius of gyration calculated from the X-ray structural coordinates with the radius of gyration calculated from the corresponding NMR structure. (B) A comparison of the radius of gyration calculated from the X-ray (square) and NMR (circle) structural coordinates with the predicted radius of gyration based on the number of residues in the protein. (C) A comparison of the radius of gyration calculated from the X-ray (circle) and NMR (square) structural coordinates with the experimental radius of gyration reported in the literature. The four proteins with extremely large deviations between the structural and experimental Rg were excluded from the graph for clarity. The line corresponding to y = x is included in each graph.
References
- (35) X. Huang, F. J. Moy, and R. Powers* (2000) "Evaluation of the Utility of NMR Structures Determined from Minimal NOE Based Restraints for Structure Based Drug Design, using MMP-1 as an Example", Biochemistry, 39(44):13365-13375. PMID11063573.
- (39) X. Huang and R. Powers* (2001) "Validity of Using the Radius of Gyration as a Constraint in NMR Protein Structure Determination", J. Amer. Chem. Soc., 123(16):3834-3835. PMID11457122.