"That is why scientists persist in their investigations, why we
struggle so desperately for every bit of knowledge, stay up nights
seeking the answer to a problem, climb steepest obstacles to the
next fragment of understanding, to finally reach that joyous moment
of the kick in the discovery, which is part of the pleasure of
finding things out."
- Richard P. Feynman (Nobel Prize in Physics, 1965)








Nuclear Magnetic Resonance (NMR) spectroscopy has proven itself an extremely versatile tool for exploring a variety of biological problems. NMR has been successfully used to determine the structure of numerous biomolecules, investigate biomolecular interactions, screen for potential drugs, determine structures of drug-complexes, and more recently as an important tool of metabolomics and systems biology. Metabolomics is used to monitor in vivo drug activity and to analyze biofluids (blood, urine, etc.) for drug toxicity, disease biomarkers, and personalized medicine. As a result, NMR is widely used in the pharmaceutical industry to aid in drug discovery efforts. Our research interest is focused on the application and development of NMR methodologies to improve the success rate and efficiency of drug discovery. This effort includes the development of NMR- and MS-based methods for metabolomics; and the application of NMR and bioinformatics to understand the structure, function and evolution of novel proteins and their corresponding therapeutic utility in structure-based drug design programs.
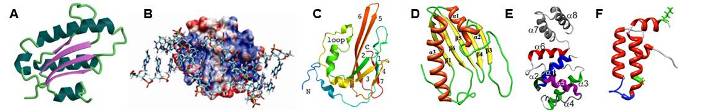
Examples of some of our recent NMR protein structures. (A) Archaeglobus fulgidis peptidyl-tRNA hydrolase, (B) Pseudomonas putida protein PpPutA45 and its DNA complex, (C) Pseudomonas aeruginosa protein PA1324, (D) protein YndB from Bacillus subtilis, (E) Staphylococcus aureus C-terminal domain of primase, and (F) J-domain of human DNAJA1.
An emerging field for NMR has been its application in metabolomics. One means of analyzing the state of a biological system is achieved by monitoring the metabolome - all the metabolites present in a cell, tissue, organ or organism . Correspondingly, metabolomics is the study of the changes in the concentration and identity of these metabolites that results from environmental or genetic stress from a disease state or drug treatment. In essence, the metabolome provides a chemical fingerprint or signature that uniquely defines the state of the system. Metabolomics has a significant advantage over genomics and proteomics since observed changes in the metabolome are directly coupled with changes in protein activity - metabolites are the end products of enzyme and protein activity. A simple change in the expression level of a gene or protein does not necessarily correlate directly with a change in activity level. Thus, metabolites are more proximal to a phenotype or disease, where NMR and MS have been routinely used to monitor these changes in the metabolome.
We have applied our metabolomics methodologies to monitor the in vivo effect of D-cycloserine on mycobacteria, the activity of 8-Azaxanthine in Aspergillus nidulans, to identify the in vivo mechanisms of action of antibiotics for tuberculosis and the discovery of novel antibiotics, to investigate gemcitabine resistance and the role of MUC1 in pancreatic cancer, to investigate the use of ketone bodies to diminish pancreatic cancer cachexia, to investigate the molecular mechanism of pesticides to Parkinson`s disease, for the discovery of disease biomarkers for Multiple Sclerosis, and the investigation of survivability, adaptability, and biofilm formation among other cellular processes related to Staphylococcus epidermis and S. aureus infections.
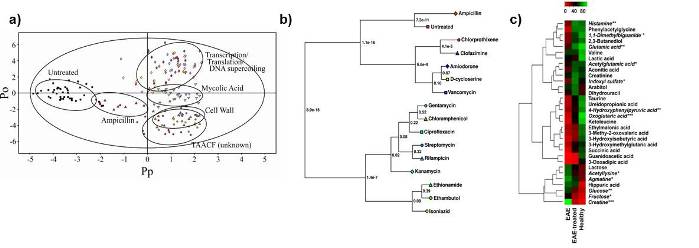
Examples of some of the typical approaches used in our lab to analyze NMR metabolomics data. (A) an OPLS-DA scores plot demonstrating the relative differences between classes. In this case, the different in vivo activity of TB drugs. (B) A metabolomics tree-diagram from our PCA/PLS-DA utilities for the statistical analysis of clustering patterns in the scores plot from A. (C) Heatmap demonstrating the relative changes in metabolite concentrations associated with Multiple Sclerosis.
We have developed and implemented NMR- and MS-based methods to monitor changes in the metabolome as a tool for systems biology, drug discovery and, for the discovery of disease biomarkers. This effort includes optimization of protocols for the preparation of metabolomics samples, development of methodologies (PCA/PLS-DA utilities) to determine the statistical significance of cluster separation in principal component analysis (PCA) orthogonal projection to latent structures-discriminate analysis (OPLS-DA), and partial least squares (PLS) scores plot, an investigation into the negative impact of noise on multivariate statistical analysis, an algorithm to simultaneously correct for phase errors and normalize NMR spectra to reduce within group variability, a deterministic nonuniform sampling (NUS) method to accelerate spectral acquisition of multidimensional NMR datasets with minimized sampling artifacts, an adaptive intelligent binning algorithm for dimensionality reduction of multidimensional NMR datasets, a software platform (MVAPACK) that centralizes and standardizes all of the processing, modeling and validation of NMR metabolomics datasets, and recent efforts in integrating both MS and NMR datasets for the analysis of metabolomics. We are actively pursuing other NMR and MS techniques to improve the coverage of the metabolome, and improve the accuracy and speed of metabolite and pathway identification. This includes enabling 2D NMR methods and ion mobility spectrometry-mass spectrometry to be routinely used in metabolomics and identify metabolites, while eliminating the need for separation techniques that confound the interpretation and diminish accuracy.
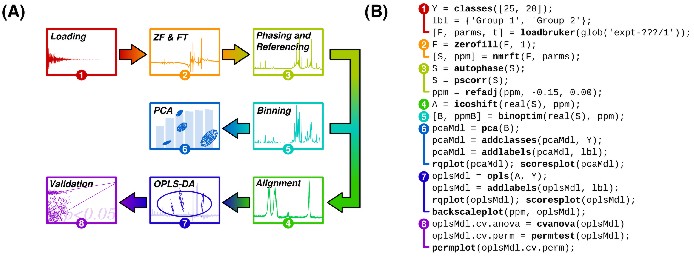
An example NMR metabolic fingerprinting data handling flow diagram (A) and its associated MVAPACK commands (B). This minimalistic data handling script is a simple starting point for using MVAPACK; much more flexibility and functionality are present in the software than can be shown here. All functions in boldface are provided in MVAPACK.
Based on the success of the Human Genome project and the Structural Genomics initiatives, thousands of functionally uncharacterized proteins have been identified. Sequence and structural similarity techniques may provide functional information for, at most, half of these proteins. To functionally annotate all known proteins will require the development of new techniques and methodology to rapidly determine protein structures and evaluate protein function. By combining the strengths of NMR, mass spectrometry and molecular modeling, rapid approaches to solving both protein and protein-ligand complexes are being explored in our group. Additionally, by modifying NMR screening techniques utilized to identify drug candidates, we are exploring the ability to quickly ascertain initial functional information for functionally uncharacterized proteins that have emerged from the Structural Genomics effort. Toward this end, we have developed the FAST-NMR technology that incorporates structural biology, ligand affinity screens and bioinformatics. Thus, the development of bioinformatics software and second generation databases are also an integral component of our research efforts. The CPASS software and database was developed to facilitate protein functional annotation through the sequence and structure comparison of ligand-binding sites. Our CPASS software has recently been demonstrated to be a valuable approach to investigate the functional evolution of proteins.
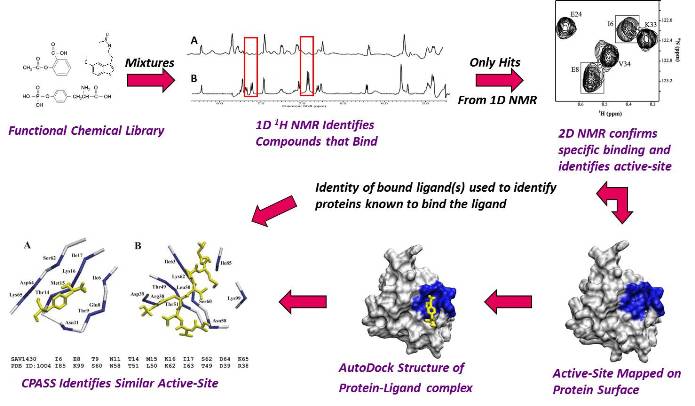
Flow Diagram of Our FAST-NMR Functional Annotation Methodology.