B. subtilis Acyl Carrier Protein (ACP)
Acyl carrier protein (ACP) is a fundamental component of fatty acid
biosynthesis where the fatty acid chain is elongated by the fatty acid
synthetase system while attached to the 4'-phosphopantetheine prosthetic
group (4'-PP) of ACP. Activation of ACP is mediated by holo-acyl carrier
protein synthase (ACPS) where ACPS transfers the 4'-PP moiety from coenzyme
A (CoA) to Ser-36 of apo-ACP. Both ACP and ACPS have been identified as
essential for E. coli viability and potential targets for development
of antibiotics.
Available FilesRelated Publications
|
A. fulgidis Peptidyl-tRNA Hydrolase (NESG AF2095, GR4)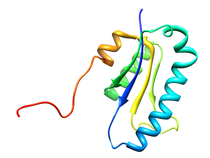
The thermophilic archaea Archaeglobus fulgidis AF2095 protein is an example
of a protein of unknown biological function targeted for structural analysis
by the Northeast Structural Genomics Consortium (NESG), which also belongs
to the previously unannotated Pfam family UPF0099. Comparison of the 3D NMR
structure of A. fulgidis AF2095 and the 1.95 angstrom X-ray crystal
structure of the functionally-unannotated thermophilic archaea
T. acidophilum protein TA0108 that has recently been deposited in the
Protein Database (PDB: 1rlk) with the human Pth2 X-ray structure suggest
that these proteins are also Pth2 enzymes.
Available FilesRelated Publications
|
S. aureus C-terminal domain of primase (DnaG)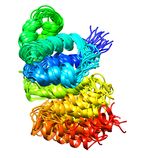
The protein-protein interaction between the C-terminal domain (CTD) of primase (DnaG)
and N-terminal domain of helicase (DnaB) is essential for DNA synthesis during
bacterial DNA replication. This interaction is conserved in all bacteria, and
distinctly different from that of eukaryotes making it an attractive antibiotic
target. To develop this interface as an antibiotic target and ligand-binding
site, we determined the solution structure and dynamics analysis of DnaG CTD
from Staphylococcus aureus. Its structure is more similar to the one from
Geobacillus stearothermophilus than from Escherichia coli, consistent
with a structural divergence between Firmicutes and Proteobacteria. The greatest
divergence lies in the final two α-helices that form the C2 subdomain. NMR
measurements indicate that the two subdomains are hold rigidly with respect
to one another and that all relative movement is focused on a single residue
N564 in S. aureus. The larger subdomain has regions undergoing rapid folding
and unfolding but the C2 subdomain has a very high order. The model that emerges
from this analysis is that 5 residues in the inflexible C2 subdomain make contact
with one of a pair of DnaB N-terminal domains. Next two residues in the partially
unfolded C1 subdomain make contact with the other DnaB NTD in a way that stimulates
ATPase activity. Since C2 subdomain-NTD interaction is species-dependent, it
is the preferred target area.
Available FilesRelated Publications
|
Human DNAJ Homologue Subfamily A Member 1 (DNAJA1, NESG HR30991)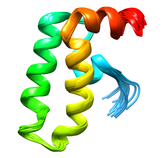
The human protein DnaJ homolog subfamily A member 1 (DNAJA1) was previously
shown to be down-regulated five-fold in pancreatic cancer cells, and has been
targeted as a biomarker for pancreatic cancer. But, little is known about the
specific biological function for DNAJA1 or the other members of the DnaJ family
encoded in the human genome. Our results suggest the overexpression of DNAJA1
suppresses the stress response capabilities of the oncogenic transcription factor,
c-Jun, and results in the diminution of cell survival. DNAJA1 likely activates a
DnaK protein by forming a complex that suppresses the JNK pathway, the
hyperphosphorylation of c-Jun, and the anti-apoptosis state found in pancreatic
cancer cells. A high-quality NMR solution structure of the J-domain of DNAJA1
combined with a bioinformatics analysis and a ligand affinity screen identifies
a potential DnaK binding site, which is also predicted to overlap with an
inhibitory binding site suggesting DNAJA1 activity is highly regulated.
Available FilesRelated Publications
|
Human Basic Fibroblast Growth Factor (FGF-2)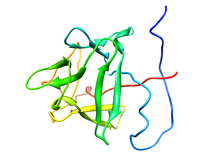
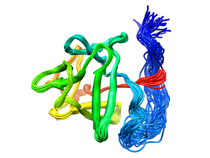
Basic fibroblast growth factor (FGF-2), a member of a protein family that includes three oncogenes (FGF-3, FGF-4 and FGF-5), exhibits angiogenic and a variety of growth and differentiation activities. Its diverse role in regulating cell growth and differentiation has suggested an involvement in wound healing, tumor growth and cancer. A common feature of the FGF family members is their high affinity toward heparin sulfate proteoglycans (HSPG). The interaction of FGF-2 with HSPG is required for high-affinity binding to its cell surface tyrosine kinase receptor (FGFR) and essential for mediating internalization and intracellular targeting through a proposed mechanism of receptor dimerization. It has been suggested that HSPG might interact directly with FGFR to facilitate the formation of a trimolecular complex and that the HSPG induced dimerization of FGF-2 may be important for receptor dimerization. Near complete 1H, 15N, 13CO, and 13C assignments, solution secondary structure, dynamics, high-resolution structure of FGF-2 and its interaction with heparin have been analyzed by NMR. A helix-like structure was observed for residues 131-136 which is part of the heparin binding site (residues 128-138). The discovery of the helix-like region in the primary heparin binding site instead of the β-strand conformation described in the x-ray structures may have important implications in understanding the nature of heparin-FGF-2 interactions. A total of seven tightly bound water molecules were found in the FGF-2 structure, two of which are located in the heparin binding site. Presented the first direct experimental evidence obtained by NMR, independently confirmed by dynamic light scattering and biological relevance established in cell-based and cell-free assays to propose a specifically oriented heparin-FGF-2 complex. Since the FGF-2-tetrasaccharide trans-dimer is inactive in receptor binding and initiation of the biological response, we conclude that the minimum active structural unit of the FGF-2-heparin complex is the properly oriented cis-dimer component of the tetramer "sandwich" motif induced by the decasaccharide. Available Files
Related Publications
|
Human Interleukin-4 (IL-4)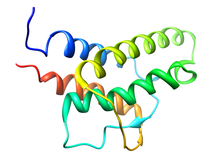
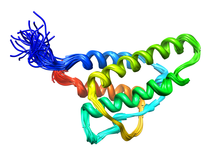
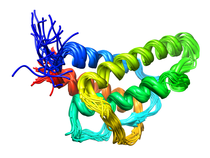
Interleukin-4 (IL-4) is one of a group of cytokines that play a central role in the control and regulation of the immune and inflamatory systems. Specific activities associated with IL-4 are the stimulation of activated B cell, T lymphocyte, thymocyte, mast cell proliferation and the induction of cytotoxic CD8+ T cells. The latter is responsible for the antitumor activity of IL-4. Renal tumor cells that secrete large doses of IL-4 can establish tumor-specific immunity toward a preexisting renal cancer. The very potent antitumor activity of IL-4 at the primary tumour site is also associated with the elicitation of a localized inflammatory infiltrate. Further, IL-4 is responsible generating and sustaining in vivo IgE and IgG1 in the T cell-dependent immune response by causing immunoglobuilin class switching of activated B cells to igE and IdG1, respectively (IL-4 overview). In order to provide a structural basis for understanding the mode of action of IL-4 and its interaction with its cell surface receptor, the NMR assignments, secondary structure and high resolution structure of IL-4 have been determined by multidimensional NMR. The structure of IL-4 is dominated by a left-handed four-helix bundle with an unusual topology comprising two overhead connections. The linker elements between the helices are formed by either long loops, small helical turns or short strands. The overall topology is remarkably similar to that of growth hormone and granulocyte-macrophage colony stimulating factor, despite the absence of any sequence homology, and substantial differences in relative lengths of the helices, the length and nature of the various connecting elements, and the pattern of disulfide bridges. These three proteins, however, bind to cell surface receptors belonging to the same hematopoietic superfamily, which suggests that interleukin-4 may interact with its receptor in an analogous manner to that observed in the crystal structure of the growth hormone-extracellular receptor complex. Available Files
Related Publications
|
Human Interleukin-13 (IL-13)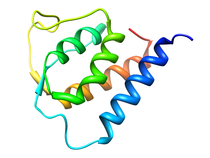
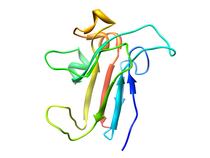
Interleukin-13 has been implicated as a key factor in asthma, allergy,
atopy and inflammatory response establishing the protein as a valuable
therapeutic target. IL-13 is produced by activated T cells and promotes
B cell proliferation, induces B cells to produce IgE, down regulates the
production of proinflamatory cytokines, increases expression of VCAM-1 on
endothelial cells, enhances the expression of class II MHC antigens and
various adhesion molecules on monocytes. IL-13 mediates these functions
through an interaction with its receptor on hematopoietic and other cell
types, but currently no functional receptors have been identified on T
cells. The signaling human IL-13 receptor (IL-13R) is a heterodimer
composed of the interleukin-4 receptor a chain (IL-4Ra) and the IL-13
binding chain. The association of IL-13 with its receptor induces the
activation of STAT6 (signal transducer and activation of transcription
6) and Janus-family kinase (JAK1, JAK2, TYK2) through a binding
interaction with the IL-4Ra chain.
Available Files
Related Publications
|
Human Class-I Histocompatibility Antigen-peptide Complexes (mAb TP25.99)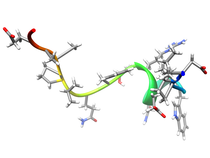
Human Class I histocompatibility antigen-peptide complexes initiate the cascade that results in the rejection of foreign tissue transplants. T cell receptor molecules bind the a1 and a2 domains of the HLA heavy chain in the antigen-peptide complex while the CD8 co-receptor binds the a3 domain to initiate a response. The anti-HLA Class I monoclonal antibody (mAb) TP25.99 has an unusual specificity as it recognizes determinants expressed on b2-m associated and b2-m free HLA Class I heavy chains. In the companion paper using phage display peptide libraries (Desai et al., 1998) we reported the identification of a cyclic and a linear peptide reacting with mAb TP25.99. The nineteen residue linear synthetic peptide sequence (X19) contains a stretch homologous to residues 239-242, 245 and 246 of HLA Class I heavy chains. The twelve residue cyclic peptide (LX-8) contains a stretch homologous to residues 194-198 of HLA Class I heavy chains. Analysis by two-dimensional transfer NOE spectroscopy of the induced solution structure of the X19 and LX-8 peptides in the presence of mAb TP25.99 showed that in spite of the lack of sequence homology, the two peptides adopt a similar structural motif. This motif corresponds to a short helical segment followed by a tight turn, reminiscent of the determinant loop region (residues 194-198) on b2-m associated HLA Class I heavy chains. An atomic rms distribution between the backbone atoms of X19 (residues 4-11) and LX-8 (residues 10-3) is 1.06 angstroms. The structural similarity between the X19 and LX-8 peptides and the lack of sequence homology suggests that mAb TP25.99 predominantly recognizes a structural motif instead of a consensus sequence. These results are also consistent with the observation that the association of HLA Class I heavy chains with b2-m modifies drastically their conformation and causes a change in their antigenic profile. Available Files
Related Publications
|
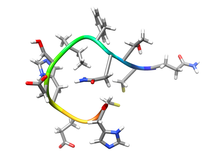
Human Fibroblast Collagenase (MMP-1)
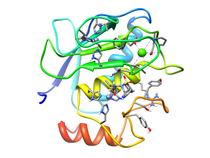
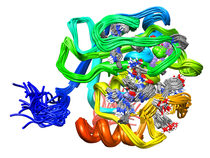
Fibroblast collagenase (MMP-1) is a member of the matrix
metalloproteinase (MMP) family which include the collagenases, stromelysins
and gelatinases. These enzymes require zinc and calcium for activity and
are modular with both propeptide and catalytic domains being common to the
entire family. The design of inhibitors of various MMPs for use as
therapeutic agents in the treatment of arthritis and cancer has been an
exceptionally active area of research. The MMPs are involved in the
degradation of the extracellular matrix that is associated with normal
tissue remodeling and, as result, MMP expression and activity is highly
controlled by either specific inhibitors (tissue inhibitor of
metalloendoproteases - TIMP), by cleavage of the inactive proenzyme or by
transcription induction or suppression. A number of biochemical stimuli
including cytokines, hormones, oncogene products and tumor promoters effect
the synthesis and activation of MMPs. The apparent loss in this regulation
can result in the pathological destruction of connective tissue and an
ensuing disease state. The MMP family consists of more than 25 enzymes,
and it has been postulated that the toxicity demonstrated by many MMP
inhibitors in clinical trials may result from non-specific inhibition.
Thus, the current approach relies on structure-based design of inhibitors
of specific MMPs, where selectivity against MMP-1 may be a desirable
trait. The extensive structural data available for the MMPs has enabled
the identification of an obvious approach for designing specificity by
taking advantage of the sequence difference and distinct size and shape of
the S1' pocket. A number of examples have been previously reported using
this approach. Nevertheless, the observed mobility of the MMP active site
may complicate the design of potentially selective inhibitors.
Available Files
Related Publications
|
Human Collagenase 3 (MMP-13)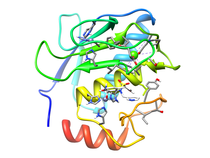
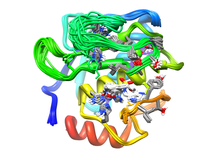
Human collagenase-3 (MMP-13) is a member of the matrix metalloproteinase (MMP) family which include the collagenases, stromelysins and gelatinases. The MMPs are involved in the degradation of the extracellular matrix which is associated with normal tissue remodeling processes such as pregnancy, wound healing, and angiogenesis. The MMPs have also demonstrated activity against cell surface and other pericellular non-matrix proteins further contributing to their cellular function. The MMP family consists of more than 25 enzymes where major discriminating factors are substrate preference (collagens, fibronectin, elastin, gelatins, etc.), domain structure and sequence alignment. The MMPs are modular proteins where a signal peptide, propeptide and catalytic domain are common to the entire family. Additional domains observed in MMP structures include fibronectin type II-like, hemopexin-like, vitronectin-like and transmembrane domains. Fundamental to the structural integrity and activity of MMPs is the presence of both zinc and calcium in the protein's structure. The active site zinc performs a critical function for both substrate binding and cleavage. Correspondingly, the design of MMP inhibitors has generally targeted the catalytic domain and active site zinc. The isolated catalytic domain maintains its general endopeptidase function but does not exhibit activity against its natural substrate. This is attributed to the absence of the hemopexin-like domain which is involved in substrate recognition and binding. The MMPs are a highly active set of targets for the design of therapeutic agents for the disease areas of arthritis and oncology. The MMPs have also been associated with multiple sclerosis, periodontitis, stroke, inflammatory bowel disease and cardiovascular disease. MMP expression and activity is highly controlled because of the degradative nature of these enzymes. The apparent loss in this regulation results in the pathological destruction of connective tissue and the ensuing disease state. MMP-13 was recently identified on the basis of differential expression in normal breast tissues and in breast carcinoma. In addition its expression has been reported in squamous cell carcinomas of the larynx, head and neck and HCS-2/8 human chondrosarcoma cells and during fetal ossification and in articular cartilage of arthritic patients. There have been a number of X-ray and NMR structures solved for the catalytic domain of MMPs complexed with a variety of inhibitors. There is a close similarity in the overall three-dimensional fold for these proteins consistent with the relatively high sequence homology (> 40%). Despite this similarity in the MMP structures there is distinct substrate specificity between these enzymes indicative of specific biological roles for the various MMPs and a corresponding association with unique disease processes. One example of this potential specificity is the over-expression of MMP-13 in breast carcinoma and MMP-1 in papillary carcinomas. Therefore the current paradigm in the development of MMP inhibitors is to design specificity into the structures of the small molecule instead of developing a broad spectrum MMP inhibitor. The rational behind this approach is that an inhibitor specific for the MMP uniquely associated with a disease process may potentially minimize toxic side effects. Comparison of the various MMP structures has identified a significant difference in the size and shape of the S1' pocket. This structural difference across the MMP family provides an obvious approach for designing specificity into potent MMP inhibitors by designing compounds that appropriately fill the available space in the S1' pocket while taking advantage of sequence differences. Available Files
Related Publications
|
Human Oncostatin M NMR-based Homology Model (OM)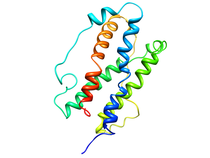
Oncostatin M (OM) is a member of cytokine family which regulates the
proliferation and differentiation of a variety of cell types and includes
interleukin-4 (IL-4), interleukin-6 (IL-6), leukemia inhibitory factor
(LIF), and granulocyte-colony stimulating factor (G-CSF). This family of
proteins adopt a four helix bundle fold with up-up-down-down topology and
contain intramolecular disulfide bonds. The shared functions of these
proteins are mediated through the interaction of the signal transducing
membrane glycoprotein, gp130, with the extracellular domains of these
cytokine receptors. Additional biological activities attributed to OM
include the inhibition of several tumor cell lines, inhibition of
embryonic stem cell differentiation a regulator of endothelial cells and
the growth stimulation of several fibroblast cell lines. OM has also been
shown to be a mitogen for AIDS-related Kaposi's sarcoma cells, where it
functions as an autocrine growth factor.
Available FilesRelated Publications
|
Pseudomonas putida Proline Utilization A, DNA-binding Domain (PutA)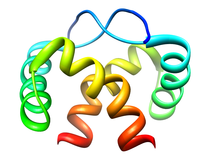

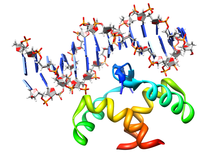
Proline utilization A (PutA) is a multifunctional enzyme that allows
Gram-negative bacteria, such as Escherichia coli and Pseudomonas
putida, to utilize proline as a carbon and nitrogen source. PutA
converts proline into glutamate in a two-step process that requires a
proline dehydrogenase domain (PRODH) and a
D1-pyrroline-5-carboxylate (P5C) dehydrogenase
domain (P5CDH). PRODH and P5CDH are separate enzymes in Gram-positive
bacteria, archea, and eukaryotes. Proline is first oxidized to P5C coupled
with the reduction of the FAD cofactor by the PutA PRODH domain. The
reduced FADH2 cofactor transfers the electrons to the electron transport
chain system in the cytoplasmic membrane. The P5CDH domain then catalyzes
the NAD+-dependant oxidation of P5C to glutamate.
Available Files
Related Publications
|
Human Regulators of G-Protein Signaling (RGS4)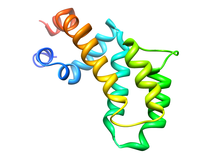
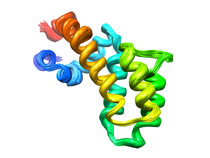
An ubiquitous component of signal transduction pathways is a heterotrimeric
guanine nucleotide-binding protein (G-protein) coupled to a cell surface
receptor. G-proteins relay signals initiated by photons, odorants, and a
number of hormones and neurotransmitters where a variety of diseases are
caused by defects in G-protein activity. The structure of the G-protein is
composed of an a-subunit (Ga) that is associated with both the intracellular
carboxy terminal tail of a seven-helical transmembrane receptor and weakly
bound to a dimer (Gbg) of a b-subunit tightly bound to a g-subunit.
G-proteins transfer signals from more than 1000 receptors where various Ga
subtypes regulate a variety of distinct downstream signaling pathways and
the guanine nucleotide binding and GTPase function within the Ga domain
regulates the activity of G-proteins.
Available Files
Related Publications
|
Human Tumor Necrosis Factor Receptor-1 (TNFR-1) Death Domain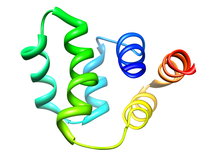
Activation of the tumor necrosis factor receptor-1 (TNFR-1) by the ligand
TNF initiates two major intracellular signalling pathways that lead to the
activation of the transcription factor NFkB and the induction of cell
death, which also elicits a variety of biological responses, including
antiviral activity, cytotoxicity, and modulation of gene expression.
TNFR-1 through trimerization is induced by the binding of TNFa
(cachectin) or TNFb (lymphotoxin a) trimers, inducing association of its
intracellular death domain (DD). The trimerization of TNFR-1 alllows for
the recruitment of an adaptor protein named TNFR-asociated death doman
protein (TRADD) through death domain-death domain interaction. TRADD
recruits the signaling molecules TNFR-associated factor-2 (TRAF-2) through
interactions with the N-terminal domain, Fas-associated death domain
protein (FADD) through death domain interactions, and the receptor
interacting protein (RIP) through death domain interactions to form the
TNFR-1 signaling complex. Based on similarities in their cystein-rich
extracellular domains, TNFR-1 and TNFR-2 belong to a receptor
superfamily, which besides a number of death inducing receptors, includes
CD40 and the low-affinity nerve growth factor receptor. Although most cell
types express both TNF receptors, TNFR-1 appears to play a predominant
role in the induction of gene expression and induction of cell death by
TNFa.
Available FilesRelated Publications
|
B. subtilis AHSA1 Protein (NESG SR211, YndB)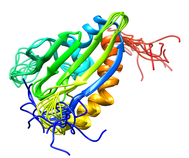
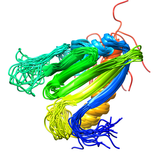
The Bacillus subtilis YndB protein is a protein of unknown biological
function targeted for structural analysis by the Northeast Structural
Genomics Consortium (NESG; http://www.nesg.org; NESG target: SR211).
The YndB protein was originally assigned as a START domain protein based on
the high sequence similarity to B. cereus BC4709 and B. halodurans BH1534,
which were assigned to START domains based on common structural features.
Instead, SCOP and Pfam databases have suggested that YndB belongs to the
closely related AHSA1 subfamily. Also, YndB, BC4709, and BH1534 do not have
the additional N-terminal β-strands and the additional α-helix that are
characteristic of a START domain structure. Similarly, a BLASTP sequence
alignment search indicates YndB is more appropriately assigned as a member
of AHSA1. The functions for prokaryotic AHSA1 family members
are typically classified as either a general stress protein or a conserved
putative protein of unknown function.
Available Files
Related Publications
|
E. coli Cell Division Protein (ZipA)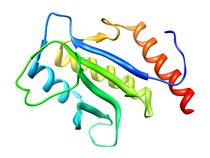
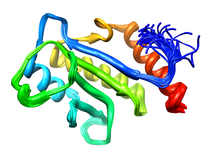
ZipA is essential for cell division and viability in E. coli.
Experiments with the green fluorescent protein fusion of ZipA have
demonstrated that only prior localization of FtsZ is required for
localization of ZipA to midcell. Changes in the relative abundance of
ZipA in the cell, either by depletion or over-expression, result in
filamentation. The morphology of these filaments resembles those observed
for cells in which FtsZ has been depleted. This is consistent with the
observation that both ZipA and FtsZ are involved at the very early stage
of cell division.
Available Files
Related Publications
|